فنیل کتونوریا - علائم کلاسیک انتقال ارثی و رژیم درمانی
مطالب
بیماری هایی که وقوع آنها مربوط به نقص دستگاه ژنتیکی سلولی - فنیل کتونوری است - در لیست کوچکی از بیماری های ارثی قابل درمان قرار می گیرند..پیشگام این بیماری یک پزشک نروژی IA Felling بود ، بعداً کشف شد که یک ژن واحد به نام ژن فنیل آلانین هیدروکسیلاز (بازوی طولانی کروموزوم دوازدهم حاوی حداکثر 4.5 درصد از کل مواد DNA سلولی) مسئول توسعه و سیر بیماری است.نقص ارثی منجر به غیرفعال سازی جزئی یا کامل آنزیم کبد فنیل آلانین-4-هیدروکسیلاز می شود.
نحوه تشخیص بیماری فنیل کتونورییا
بیماری فنیل کتونوریای ارثی (PKU) منجر به مسمومیت مزمن بدن با مواد سمی می شود که در نتیجه اختلال در متابولیسم و اسید آمینه ایجاد می شود.هیدروکسیلاسیون فنیل آلانین.مسمومیت دائمی باعث آسیب به سیستم عصبی مرکزی (CNS) می شود ، تجلی آن کاهش تدریجی در هوش (الیگوفرنی فنیل پیروویک) است.
بیماری فلجینگ در تجمع بیش از حد در بدن فنیل آلانین و محصولات متابولیسم نادرست آن آشکار می شود.سایر عوامل در ایجاد فنیل کتونوری شامل اختلال در حمل و نقل اسید آمینه در سراسر سد خونی مغزی ، تعداد کم انتقال دهنده عصبی (سروتونین ، هیستامین ، دوپامین) می باشد.در صورت عدم درمان به موقع بیماری منجر به عقب ماندگی ذهنی می شود و می تواند باعث مرگ کودک شود.

مکانیسم توسعه بیماری
علت اختلالات ژنی بلوک متابولیکی است که از تشکیل فنیل آلانین-4-هیدروکسیلاز جلوگیری می کند (آنزیمی ،که وظیفه تبدیل اسید آمینه فنیل آلانین به تیروزین را دارد).تیروزین اسید آمینه پروتئینزا بخشی جدایی ناپذیر از پروتئین ها و رنگدانه ملانین است ، بنابراین یک عنصر ضروری برای عملکرد کلیه سیستم های بدن است و فقدان آن منجر به فریموپاتی می شود.
مهار تشکیل متابولیت ناشی از غیرفعال شدن جهش آنزیم ، فعال سازی مسیرهای متابولیکی جانبی فنیل آلانین است.آلفا آمینه آروماتیک معطر ، به عنوان یک نتیجه از نقص فرآیندهای متابولیکی ، مشتقات سمی را تشکیل می دهد که در شرایط عادی تشکیل نمی شود:
- اسید فنیل پیروویک (فنیل پیرووات) - یک اسید چرب معطر آلفا-کتو ، تشکیل آنمنجر به اختلال در فرآیندهای عصبی و زوال عقل می شود.
- فنیل لاکتیک اسید محصولی است که در طول بازیابی اسید فنیل پیروویک تشکیل شده است.
- فنیل اتیل آمین - ترکیب اولیه انتقال دهنده های فعال بیولوژیکی پالسهای الکتروشیمیایی ، غلظت دوپامین ، آدرنالین و نوراپی نفرین را افزایش می دهد.
- ارتو فنیل استات یک ماده سمی است که باعث اختلالات متابولیک در مغز می شود.
آمار پزشکی نشان می دهد که یک ژن تغییر یافته پاتولوژیک در 2٪ از جمعیت وجود دارد ، اما به هیچ وجه خود را نشان نمی دهد.این نقص ژنتیکی فقط در صورت وجود بیماری در هر دو شریک از والدین به کودک منتقل می شود ، در حالی که شیرخوار در 50٪ موارد تبدیل به ناقل ژن جهش یافته می شود و همچنان سالم مانده است.احتمال اینکه فنیل کتونوری در نوزادان منجر به بیماری شود 25٪ است.
با چه نوع وراثتی به دست می آید
بیماری قطع فروش یک ناهنجاری ژنتیکی است که از نوع اتوزوم مغلوب به ارث رسیده است.این نوع وراثت به این معنی است که ایجاد علائم یک بیماری مادرزادی تنها هنگامی اتفاق می افتد که یک ژن معیوب از هر دو والدینی که حامل هتروزیگوس ژن تغییر یافته هستند کپی شود از کودک به ارث می رسد.
ایجاد یک بیماری مادرزادی در 99٪ موارد ناشی از جهش ژن مسئول رمزگذاری آنزیم است که سنتز فنیل آلانین-4-هیدروکسیلاز (فنیل کتونوریای کلاسیک) را فراهم می کند.حداکثر 1٪ از بیماریهای ژنتیکی مربوط به تغییرات جهشی است که در سایر ژنهای ایجادشده رخ می دهدکمبود دی هیدروپریتین ردوکتاز (PKU نوع II) یا تتراهیدروبیوپتین (PKU نوع III).
فنیل کتونوریا در کودکان
شکل کلاسیک بیماری ژنتیکی در کودکان در اکثر موارد ، از 3 تا 9 ماهگی بروز می کند و از نظر ظاهری قابل توجه است.نوزادانی که دارای یک ژن معیوب هستند ، به نظر سالم می رسند ، یک ویژگی خاص عادت (ظاهر) خاص کودک است.علائم بیان شده در 6-12 ماه پس از تولد ظاهر می شود.
PKU نوع II با این واقعیت مشخص می شود که اولین علائم بالینی 1.5 سال پس از تولد ظاهر می شود.علائم بیماری پس از تشخیص ناهنجاریهای ژنتیکی و شروع رژیم درمانی از بین نمی رود.این نوع بیماری مادرزادی غالباً منجر به یک نتیجه کشنده در 2-3 سال از زندگی کودک می شود.شایع ترین علائم PKU نوع II عبارتند از:
- اختلالات شدید روانی.
- هایپر رفلکسی؛
- اختلال در عملکرد حرکتی کلیه اندامها.
- سندرم انقباضات عضلانی کنترل نشده.
علائم بالینی تغییرات جهش در ژنهای نوع III مشابه بیماری نوع II است.کمبود تتراهیدروبیوپتین با یک مثلث از علائم خاص مشخص می شود:
- درجه بالایی از عقب ماندگی ذهنی.
- اندازه جمجمه نسبت به سایر قسمت های بدن کاملاً کاهش می یابد.
- اسپاسم عضلانی (از بین رفتن کامل تحرک اندام ممکن است).

مظاهر بیماری قطع بیماری
در آزمایشات و مشاهدات بالینی ، پیشنهاد شده است کهمشتقات سمی متابولیسم فنیل آلانین باعث کاهش توانایی فکری می شود که از نظر ماهیتی پیشرفتی است و می تواند به زوال عقل منجر شود (الیگوفرنی ، ایدیوسی).از جمله علل احتمالی اختلالات برگشت ناپذیر فعالیت مغز از معقول ترین در نظر گرفته می شود که با کاهش سطح تیروزین عدم انتقال دهنده های عصبی که باعث انتقال تکانه ها بین نورون ها می شود ، ایجاد می شود.
رابطه دقیق علت و معلولی بین بیماری های ارثی و اختلالات مغزی هنوز مشخص نشده است ، همانطور که مکانیسم توسعه به دلیل فنیل کتونوریایی از حالت های روانی مانند اکوپراکسی ، اکولالیا ، حملات شدید و تحریک پذیری است.نتایج تجزیه و تحلیل نشان می دهد که فنیل آلانین اثر سمی مستقیم بر مغز دارد ، که همچنین می تواند باعث کاهش هوش شود.
ساختار و ویژگی های فنوتیپی
با توجه به اینکه اشباع رنگدانه های پوستی و مو بستگی به میزان تیروزین در میتوکندری کبدی دارد و فنیل کتونوری منجر به توقف تبدیل فنیل آلانین می شود ، بیماران مبتلا به این بیماری ویژگی های خاصی دارند.علائم مغلوب).افزایش لحن عضلانی باعث بروز انحرافات در ساختار بدن می شود - این بیماری ناخوشایند می شود.ویژگی های بارز خارجی فنیل کتونوری شامل موارد زیر است:
- hypopigmentation - پوست روشن ، چشم آبی کم رنگ ، موهای تغییر رنگ.
- سیانوز اندام؛
- اندازه سر را کاهش داد.
- موقعیت خاص بدن - کودک هنگام تلاش برای ایستادن یا نشستن ، کودک وضعیت "خیاطی" را اتخاذ می کند (بازوها و پاها در مفاصل خم می شود).
علائمبیماری
با تشخیص به موقع ، بیماری فلجینگ با تنظیم تغذیه با موفقیت درمان می شود و رشد کودک مطابق با گروه سنی او رخ می دهد.مشکل تشخیص یک جهش ژنتیکی این است که علائم اولیه تشخیص حتی توسط یک متخصص کودکان با تجربه دشوار است.شدت علائم بیماری مادرزادی با بزرگتر شدن کودک افزایش می یابد ، زیرا مصرف غذای پروتئین به پیشرفت اختلالات CNS کمک می کند.
علائم در نوزادان
در اولین روزهای زندگی کودک ، نشانه های ناهنجاری غیرطبیعی تشخیص دشوار است - کودک به طور طبیعی رفتار می کند ، هیچ تاخیری در رشد ندارد.علائم بیماری برای اولین بار 2-6 ماه پس از تولد ظاهر می شود.والدین باید نسبت به رفتار کودک هوشیار باشند ، که با کمبود فعالیت ، بی حالی و یا برعکس اضطراب ، تحریک پذیری بیش از حد مشخص می شود.
با شروع تغذیه با شیر مادر ، نوزادان با شیر شروع به ورود به بدن نوزادان با شیر می کنند ، که یک کاتالیزور برای ظهور اولین علائم است ، که به وضوح نشان می دهد که این بیماری در حال پیشرفت است.تظاهرات بالینی خاص بیماری عبارتند از:
- استفراغ مداوم (اغلب باریک بودن مادرزادی دروازه بان در نظر گرفته می شود).
- استفراغ مکرر؛
- پاسخی به محرکهای خارجی نیست.
- دیستونی عضلانی (کاهش فشار عضله)؛
- سندرم تشنج (تشنج از صرع یا غیر صرع).
علائم در كودكان پس از 6 ماه
اگر تظاهرات بيماري ژنتيكي نباشددر 6 ماه اول پس از تولد كودك رخ داده است (یا مشاهده نشده است) ، پس از این مدت می توان به طور دقیق میزان تاخیر در رشد روانی را تعیین كرد.علائم اختلالات ژنتیکی ناشی از کمبود آنزیم در کودکان بزرگتر از شش ماه عبارتند از:
- کاهش فعالیت (تا بی تفاوتی کامل).
- عدم مهار نفس ، صندلی؛
- بوی عجیب و غریب "موش" از پوست (بوی قالب ناشی از دفع مشتقات سمی فنیل آلانین از طریق غدد عرق و ادرار است).
- از دست دادن توانایی تشخیص بصری چهره والدین.
- لایه برداری پوست؛
- درماتیت ، اگزما ، اسکلرودرمی.
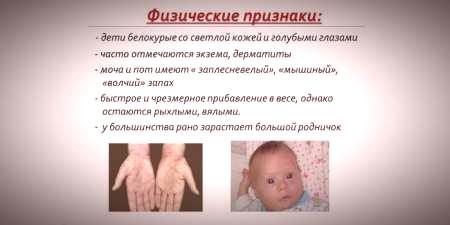
پیشرفت بیماری در غیاب درمان در کودکی
اگر ناهنجاری های رشدی در کودکی تشخیص داده نشده بود و درمان مناسب انجام نشده است ، پسبیماری شروع به پیشرفت جدی می کند و اغلب منجر به ناتوانی می شود.عدم درمان در مراحل اولیه بیماری باعث بروز علائم زیر در سن 1.5 سالگی می شود:
- میکروسفالی (کاهش اندازه مغز).
- پیش آگهی (جابجایی دندان فوقانی به جلو)؛
- دندان دیر رسیدن؛
- هیپوپلازی مینای دندان (نازک شدن یا عدم حضور کامل مینای دندان).
- تأخیر در پیشرفت زبان تا غیبت کامل گفتار؛
- 3 ، 4 درجه الیگوفرنی (عقب ماندگی ذهنی ، عقب ماندگی ذهنی)؛
- نقص مادرزادی قلب (نقص در ساختار عضله قلب ، بخش هایی از قلب ،کشتی های بزرگ)؛
- اختلالات سیستم اتونوم (آکروسیانوز ، تعریق ، افت فشار خون شریانی).
- یبوست.
علل و عوامل تحریک کننده
برای تجلی یک جهش مغلوب اتوزومی ، یک ژن معیوب باید از هر دو والدین به ارث برسد.بیماریهای ژنتیکی از این نوع در دختران و پسران تازه متولد شده با همین فرکانس اتفاق می افتد.پاتوژنز PKU ناشی از اختلال در متابولیسم فنیل آلانین است که می تواند در 3 شکل ایجاد شود.رژیم درمانی فقط در معرض فنیل کتونوریای کلاسیک نوع I قرار دارد.
اشکال آتیپیک بیماری با تنظیم تغذیه قابل درمان است.این انحرافات ناشی از کمبود تتراهیدروپتین ، دهیدروپتین ردوکتاز (به ندرت - پیروویل تتاهیدروپتین سنتاز ، گوانوزین -5-تری فسفات و موارد دیگر) می باشد.بیشتر موارد مرگ و میر در بین بیماران با نادر از PKU مشاهده می شود که تظاهرات بالینی همه اشکال این بیماری مشابه است.اگر والدین نزدیك (در ازدواجهای نزدیك) باشند ، خطر تولد نوزاد با ژن فنیل آلانین هیدروكسیلاز جهش یافته افزایش می یابد.
تشخیص
در صورت وجود مشکوک به اختلالات ژنتیکی ، تشخیص بر اساس مجموعه ای از داده های بدست آمده از مطالعه تاریخ پزشکی انجام می شود - داده های شجره نامه ، نتایج مطالعات ژنتیکی بالینی و پزشکی.برای شناسایی به موقع بیماریهای مادرزادی (PKU ، فیبروز کیستیک ، گالاکتوزمی و غیره) برنامه توده ای اجباری تهیه شدمعاینه آزمایشگاهی کلیه نوزادان (غربالگری نوزادان).
اگر والدین انتظار از ناقل ژن جهش یافته آگاهی داشته باشند ، پزشکی مدرن روش هایی برای تشخیص نقص در مرحله بارداری (تشخیص پیش از تولد جنین به روش تهاجمی) ارائه می دهد.برای تقسیم فنیل کتونوریا به انواع بر حسب شدت ، از یک طبقه بندی مشروط بر اساس سطح فنیل آلانین در یک مایع پلاسمائی خون بدون فیبرینوژن استفاده می شود:
- فنیل کتونوریای سنگین - 1200 میکرومول در لیتر.
- میانگین - 60-1200 میکرومول در لیتر.
- نور (بدون نیاز به درمان) - 480 میکرومول /لیتر.
آزمایش غربالگری
تشخیص ناهنجاری های ژنتیکی در چند مرحله اتفاق می افتد.در مرحله اول در بیمارستان زایمان ، تمام نوزادان به مدت 3-5 روز از زندگی خون گرفته می شوند (از پنج مورد) برای تحقیق.مواد روی فرم کاغذی اعمال می شوند و در آنجا تجزیه و تحلیل بیوشیمیایی به آزمایشگاه بیوشیمیایی ارسال می شوند.در مرحله دوم تست غربالگری ، غلظت غلظت طبیعی فنیل آلانین مشخص می شود.

اگر هیچ تغییر پاتولوژیکی تشخیص داده نشود ، تشخیص کامل شده و کارت کودک ثبت می شود.در صورت انحراف از هنجار ، نتایج تشخیص به پزشک متخصص اطفال فرستاده می شود تا از معاینه دقیق تر نمونه خون خون نوزاد اطمینان حاصل شود.سلامت کودک به اجرای به موقع و دقیق کلیه اقدامات لازم برای تشخیص انحرافات بستگی دارد.اگر تشخیص بعد از غربالگری مكرر تأیید شود ، والدین كودك انجام می دهندبرای معالجه به کلینیک ژنتیک کودکان ارسال شد.
تجزیه و تحلیل ها و مطالعات برای تأیید تشخیص
تشخیص مجدد هنگام تشخیص در هنگام آزمایش غربالگری اولیه ناهنجاری ها با ارسال مجدد آزمایش ها انجام می شود.علاوه بر تعیین مقدار فنیل آلانین در خون به روشهای تشخیص PKU در کودکان و بزرگسالان شامل:
- آزمایش قطع فروش - تعیین فنیل پیروویک اسید در ادرار با اضافه کردن کلرید آهن به ماده بیولوژیکی (رنگ آمیزی به رنگ آبی-سبز).
- تست گوتری - ارزیابی میزان پاسخ میکروارگانیسم ها به متابولیسم یا آنزیم های موجود در خون بیمار؛
- کروماتوگرافی - بررسی خصوصیات شیمیایی مواد توزیع شده بین دو مرحله.
- فلوریمومتری - تابش ماده بیولوژیکی توسط تابش تک رنگ برای تعیین غلظت مواد موجود در آن.
- الکتروانسفالوگرافی - تشخیص فعالیت الکتریکی مغز؛
- تصویربرداری با رزونانس مغناطیسی تحریک هسته های اتمی سلول ها توسط امواج الکترومغناطیسی و اندازه گیری پاسخ آنها است.
درمان فنیل کتونوریای کلاسیک
در قلب فنیل کتونوریا درمانی محدودیت در مصرف محصولاتی است که منبع پروتئین های حیوانی و گیاهی هستند.تنها روش درمانی موفق ، رژیم درمانی است که میزان کفایت آن با محتوای فنیل آلانین موجود در سرم ارزیابی می شود.حداکثر میزان اسیدهای آمینه در بیماران گروههای سنی مختلفاست:
- در نوزادان و کودکان تا 3 سال - تا 242 میکرومول در لیتر ؛
- در کودکان پیش دبستانی تا 360 میکرومول در لیتر؛
- در بیماران 7 تا 14 ساله - تا 480 میکرومول در لیتر؛
- در نوجوانان - حداکثر 600 میکرومول در لیتر.
اثربخشی رژیم بستگی به این دارد که در چه مرحله از بیماری رژیم اصلاح شده است.در تشخیص اولیه آسیب شناسی مادرزادی ، رژیم درمانی از هفته هشتم زندگی تجویز می شود (پس از این مدت تغییرات برگشت ناپذیر از قبل شروع می شود).عدم وجود اقدامات به موقع منجر به بروز عوارض و كاهش سطح هوش تا 4 نقطه به مدت 1 ماه از زمان تولد تا شروع درمان می شود.

با توجه به اینکه رژیم درمانی فنیل کتنوریا امکان خروج کامل از رژیم غذایی پروتئین حیوانی را فراهم می کند ، نیاز به استفاده از منابع دیگر اسیدهای آمینه ضروری و همچنین ویتامین های گروه B ، کلسیم -و ترکیبات معدنی حاوی فسفرمحصولاتی که به عنوان مکمل های پروتئین مورد استفاده قرار می گیرند عبارتند از:
- هیدرولیز پروتئین (آمیژن ، آمینازول ، فیبرینوزول).
- حاوی مخلوط های فنیل آلانین اشباع شده با اسیدهای آمینه ضروری نیستند - تترافن ، بدون فنیل.
علاوه بر اقدامات درمانی برای از بین بردن علت اختلال در عملکرد بدن ، باید درمان علامتی نیز با هدف از بین بردن نقص گفتار و عادی سازی هماهنگی حرکات انجام شود.درمان پیچیده شامل روشهای فیزیوتراپی ، ماساژ ، کمک به گفتاردرمانگر ، روانشناس ، انجام تمرینات ژیمناستیک است.در برخی موارد ، همراه با رژیم درمانیاستفاده از داروهای ضد تشنج ، نوتروپیک و داروهای عروقی را نشان می دهد.
خصوصیت های درمان اشکال غیر معمول
فنیل کتونوری نوع II و III با رژیم کم پروتئین قابل درمان نیست - سطح فنیل آلانین در خون بدون تغییر باقی می ماند و محدود کردن جریان پروتئین به بدن یا علائم بالینی حتی با کاهش سطح اسید آمینه پیشرفت می کند.درمان مؤثر این اشکال از بیماری با استفاده از:
- تتراهیدروبیوپتین - عامل آنزیم تحت تأثیر انجام می شود.
- آنالوگ مصنوعی تتراهیدروبیوپتین - این مواد بهتر در سد خونی مغزی نفوذ می کنند.
- داروهای جایگزین درمانی - علت فنیل کتونوری را از بین نبرید ، اما عملکرد طبیعی بدن را پشتیبانی می کنید (لوودوپا به همراه Carbidofa ، 5-oxytryptophan ، 5-formyltetrahydrofolate).
- کبدی - محافظت از عملکرد کبد.
- ضد تشنج؛
- معرفی ژن فنیل آلانین هیدروکسیلاز به کبد یک روش تجربی است.
تغذیه و رژیم درمانی نوزادان
تغذیه با شیر مادر در سال اول زندگی یک نوزاد مبتلا به PKU مجاز است ، اما باید محدود شود.تا 6 ماه ، میزان قابل قبول فنیل آلانین 60-90 میلی گرم در هر کیلوگرم وزن کودک است (100 گرم شیر حاوی 5.6 میلی گرم فنیل آلانین است).از 3 ماه ، رژیم کودک باید به تدریج گسترش یابد و آب میوه ها و پوره ها را در آن وارد کنید.
کودکان 6 ماهه مجاز به رژیم غذایی پوره های گیاهی ، فرنی (ساگو) ، بدون پروتئین هستنداسیدبعد از 7 ماه می توان از 8 ماه به ماکارونی های کم پروتئین ، یعنی نان ، که حاوی پروتئین نیست ، به کودک ماکارونی با پروتئین کم داد.سنی که پروتئین در بدن کودک بیمار باید محدود شود ، مشخص نشده است.پزشکان هنوز درمورد امکان سنجی رژیم درمانی درمانی مادام العمر بحث می کنند ، اما آنها موافق هستند که رژیم غذایی حداقل 18 سال باید دنبال شود.
فنیل کتونوریا که در زن تشخیص داده می شود دلیلی برای امتناع از تولد فرزند نیست.مادران انتظار با PKU برای جلوگیری از آسیب جنین در دوران بارداری و جلوگیری از عوارض احتمالی ، لازم است رژیم غذایی را با رژیم محدود شده با فنیل آلانین (در زمان زایمان) رعایت کنید (حداکثر 242 میکرومول در لیتر).
مخلوط عاری از لاکتوز برای نوزادان
رژیم غذایی برای فنیل کتونوری مبتنی بر کاهش قابل توجه مقدار پروتئین طبیعی در رژیم غذایی روزانه است ، اما بدن نوزاد در صورت وجود عناصر ضروری لازم نمی تواند رشد کند.برای رفع نیاز کودک به پروتئین ، از مخلوط های اسید آمینه عاری از لاکتوز استفاده می شود که طبق قانون روسیه باید بصورت رایگان تهیه شود.
تحمل نوزاد به فنیل آلانین به سرعت در طول سال اول زندگی تغییر می کند ، بنابراین باید غلظت آن در خون کودک کنترل شود و رژیم غذایی تنظیم شود.این مخلوط ها برای گروه های سنی خاص طراحی شده است:
- نوزادان زیر یک سال ، Afenilak 15 ، Analog-SP ، PKU-1 ، PKU-mix ، PKU Anamix تجویز می شوند.
- کودکان بزرگتر از 1 سالسال ، انتصاب غنی شده با ویتامین ها و مواد معدنی با پروتئین زیاد - PKU Prima ، P-AM Universal ، PKU-1 ، PKU-2 ، Maximeid XP ، Maximum XP.

غذاهای مکمل پروتئین
یکی از مهمترین مؤلفه های رژیم غذایی برای فنیل کتونوری ، محصولات مبتنی بر نشاسته پروتئین کم است.این مکمل ها حاوی کازئین هیدرولیز ، تریپتوفان ، تیروزین ، متیونین ، نیتروژن هستند و نیاز روزانه کودک به پروتئین مورد نیاز برای رشد و نمو طبیعی را تأمین می کنند.غذاهای تخصصی که فقدان مواد معدنی ضروری و اسیدهای آمینه را در هنگام عدم رژیم در بدن پر می کنند عبارتند از:
- Berlofen؛
- زیمورگان؛
- مینافن؛
- آپونتی.
رژیم غذایی برای کودکان پیش دبستانی و دانش آموزان مدرسه
با تنظیم بدن به فنیل آلانین ، کودکان از 5 سالگی به تدریج می توانند محدودیت های غذایی خود را کاهش دهند.گسترش رژیم با ورود غلات ، محصولات لبنی ، فرآورده های گوشتی رخ می دهد.دانش آموزان ارشد قبلاً تحمل بالایی به فنیل آلانین دارند ، بنابراین در این سن شما می توانید ضمن نظارت بر پاسخ به هرگونه تغییر در تغذیه ، همچنان به گسترش رژیم غذایی ادامه دهید.از روشهای زیر برای کنترل وضعیت کودک استفاده می شود:
- ارزیابی پارامترهای عصبی ، وضعیت روانی؛
- کنترل عملکرد الکتروانسفالوگرام؛
- تعیین سطح فنیل آلانین.
گروه های غذایی با PKU
رژیم غذایی بیماران مبتلا به PKU ، همراه با پروتئین کممحصولات نشاسته ای و مخلوط های دارویی نیز شامل محصولاتی با منشا طبیعی هستند.هنگام ترسیم منو ، میزان پروتئین مصرفی باید به طور واضح محاسبه شود و از دوز توصیه شده توسط پزشک نباید تجاوز کرد.برای از بین بردن اثرات سمی روی بدن ، 3 لیست از محصولات تهیه شده است که شامل موارد ممنوع (قرمز) ، توصیه نمی شود (نارنجی) و مجاز (سبز) باشد.
فهرست قرمز
فنیل کتونورییا در زمینه عدم وجود آنزیمی که به تیروزین فنیل آلانین تبدیل می شود ، توسعه می یابد ، بنابراین پروتئین بالایی پایه ای برای لیست محصولات موجود در لیست ممنوع (قرمز) است.موارد موجود در این لیست باید رژیم غذایی بیمار مبتلا به PKU را به طور کامل حذف کند:
- گوشت؛
- اندام داخلی حیوانات ، محصولات جانبی؛
- سوسیس ، کالباس؛
- غذاهای دریایی (از جمله ماهی)؛
- تخم مرغ از همه پرندگان؛
- لبنیات؛
- آجیل؛
- حبوبات و غلات؛
- محصولات سویا؛
- ظروف حاوی ژلاتین؛
- شیرینی پزی؛
- آسپارتام.
فهرست نارنجی
محصولاتی که برای کودک مبتلا به PKU تشخیص داده می شوند ، در لیست نارنجی ها قرار می گیرند.گنجاندن در رژیم غذایی اقلام موجود در این لیست قابل قبول است اما با تعداد بسیار محدود.اگرچه این محصولات حاوی پروتئین زیادی نیستند ، اما همچنین می توانند سطح فنیل آلانین را افزایش دهند ، بنابراین استفاده از آنها توصیه نمی شود:
- سبزیجات کنسرو؛
- ظروف سیب زمینی و برنج؛
- کلم؛
- شیر؛
- شربت.
لیست سبز
محصولات بدون پروتئین مجاز به استفاده در بیماران با تشخیص فنیل کتونورییا بدون محدودیت است.قبل از خرید اقلام موجود در لیست سبز ، لازم است ترکیب موجود در بسته را بررسی کنید و مطمئن شوید که هیچ رنگی برای آسپارتام حاوی فنیل آلانین وجود ندارد:
- میوه؛
- سبزیجات (به جز سیب زمینی و کلم)؛
- انواع توت؛
- سبزی؛
- غلات نشاسته ای (ساگو)؛
- عسل ، شکر ، مربا؛
- محصولات آرد تهیه شده از ذرت یا آرد برنج؛
- روغن ، چربی (کره ، آفتابگردان ، زیتون).
چگونه می توان سطح فنیل آلانین را در خون کنترل کرد
فنیل کتونوریا یک بیماری غیر قابل درمان است که با استفاده از رژیم غذایی و اقدامات درمانی می تواند به مرحله رکود منتقل شود.در تغییر شرایط زندگی ، اختلال در رژیم غذایی بیماری می تواند دوباره تشدید شود ، بنابراین بیماران نیاز به مشاهده مادام العمر دارند.فرایند کنترل تعیین دوره ای میزان فنیل آلانین در خون است.فراوانی سنجش به سن بیمار بستگی دارد:
- تا 3 ماه - غربالگری خون باید به صورت هفتگی انجام شود تا نتایج مداوم حاصل شود.
- از 3 ماه تا 1 سال - 1-2 بار در ماه؛
- 1 تا 3 سال - هر 2 ماه یک بار؛
- مسن تر از 3 سال - سه ماهه.

خون برای تجزیه و تحلیل 3-4 ساعت بعد از مصرف داده می شود.علاوه بر غربالگری ، پیشرفت PKU با تعیین وضعیت تغذیه ای ، رشد جسمی ، عاطفی بیمار ، سطح توانایی های فکری کنترل می شود.و توسعه زباننتایج مشاهدات ممکن است با درگیر شدن متخصصان مناسب به تشخیص دیگری نیاز داشته باشد.
Video
اطلاعات ارائه شده در این مقاله فقط برای راهنمایی است.در این مقاله نیازی به خوددرمانی نیست.فقط یک پزشک متخصص می تواند بر اساس خصوصیات فردی بیمار خاص ، تشخیص و توصیه کند.














